
This week marks the “official” release of HybPiper, the bioinformatics pipeline I’ve been working on for most of my post-doc. We published a paper on the method, and demonstrated how it can be used for phylogenetics, in this month’s Applications in Plant Sciences (open access).
This work was a collaboration between the Pleurocarpous Moss Tree of Life team and the Zerega lab at the Chicago Botanic Garden. We used draft genomic data generated from Artocarpus camansi, the wild progenitor of breadfruit, to design a set of genetic markers to be used for phylogenetics and gene family evolution analysis (for more details, see the companion paper in APPS). Using a technique known as HybSeq, high-throughput sequencing libraries are enriched for genes of interest via hybridization with short RNA probe sequences. Although a few other methods exist to tackle target enrichment data, our method is more streamlined for HybSeq data: extracting coding sequences and the flanking intron sequences for phylogenetics. In the paper we describe HybPiper and demonstrate its use on 458 genes enriched for 28 species of Artocarpus and its relatives.
Main Features
To conduct phylogenetic analysis, high-throughput DNA sequencing reads need to be re-assembled into continuous sequences. HybPiper uses several pre-existing bioinformatics tools to automate the process while maintaining an organized set of intermediate files that can aid in more detailed analysis. The main script of HybPiper has three phases:HybPiper_Infographic
Sorts reads by mapping them to target sequences, using BLASTx (protein targets) or BWA (nucleotide targets).
Assembles contigs for each gene separately.
Aligns contigs to target sequences and extracts exon (coding) sequence.
HybPiper also includes a number of scripts that can be used to extract more information from the sequencing data, including:
Coverage depth and target enrichment efficiency data, including a script for plotting a heat map of gene recovery.
Retrieval of non-coding flanking sequences (i.e. introns) either separately or together with the coding sequences (supercontigs).
Identification of putative paralogous sequences, and methods to help distinguish ancient from recent paralogs.
Process HybPiper results from many samples; for example, generation of separate FASTA files for each gene, ready for phylogenetics pipelines
For more information about HybPiper, including complete tutorials on installation, usage, and an example toy dataset, check out the GitHub page and the HybPiper wiki.
Developing HybPiper
Coming up with a consistent pipeline for processing target enrichment data was one of the primary tasks for my post-doc as part of the NSF Tree of Life grant that our team received in 2013. As part of that grant, we’ve developed over 800 markers to reconstruct the phylogeny of nearly 400 pleurocarpous mosses, so I new that the pipeline would have to be very efficient. Along the way I discovered tools such as GNU Parallel, which greatly improved the speed of mapping, sequence assembly and exon extraction. Early in the process we made the decision to develop a pipeline that could itself be a product of the grant, a tool with broad applications to address the growing use of high-throughput sequencing and target enrichment in phylogenetic systematics of non-model organisms.
Many of the features in HybPiper were suggested by several of the co-authors on the APPS paper, particularly Yang Liu, Rafael Medina, and Elliot Gardner. Twitter also played an important role: when I discovered that the assembler I was using (Velvet) was generating quite questionable assemblies, a couple people suggested SPAdes as a replacement, which has worked out great!
Naming HybPiper
Just before submitting the manuscript, my advisor called me into his office and with a very serious tone said “You need a new name for the pipeline.” One week later, we had one of the most entertaining lab meetings ever– here’s a brief look into the creative process. HybPiper brainstorm
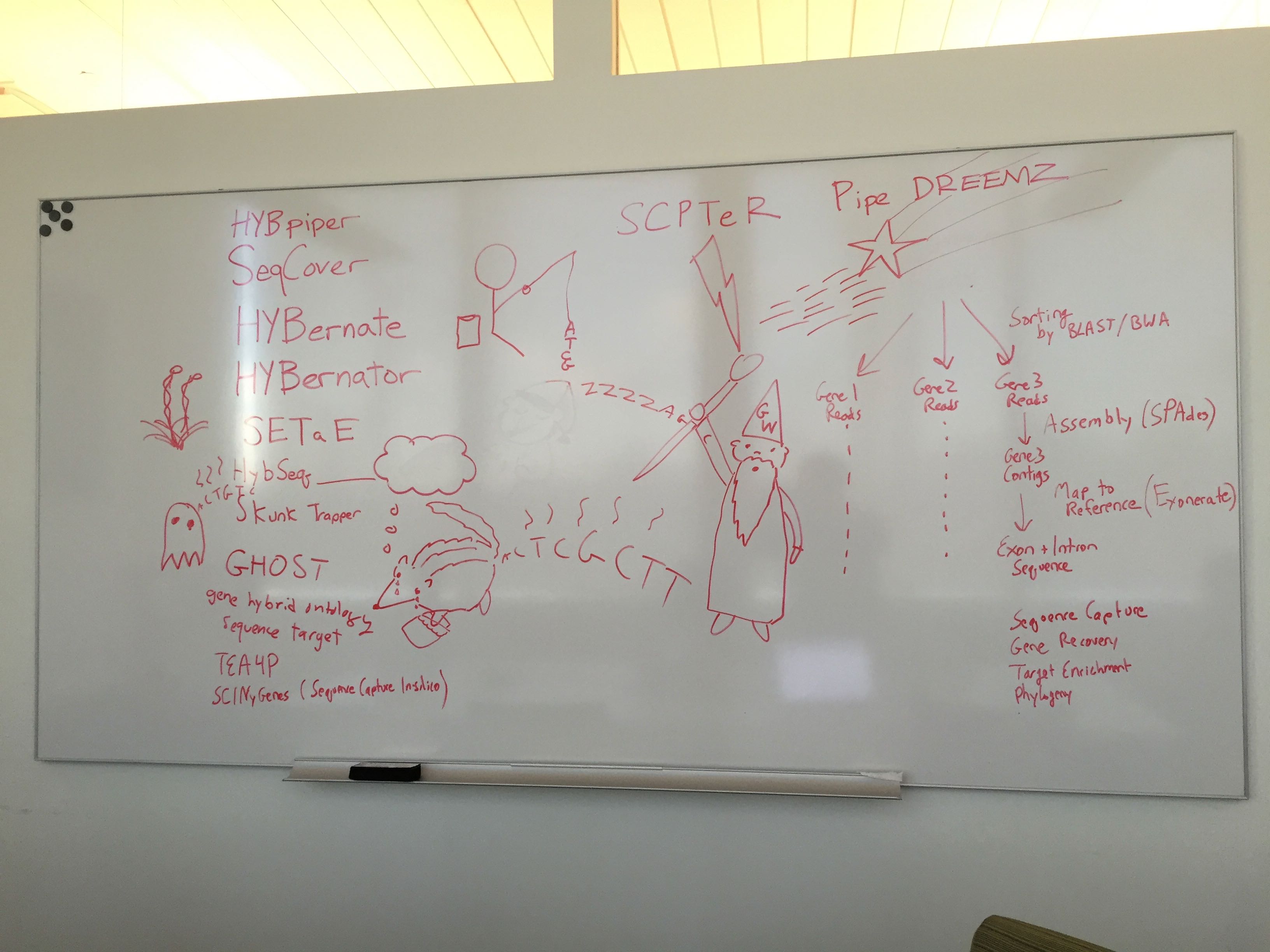
After 46 minutes and nearly calling the pipeline Skunk Trapper, we had an epiphany with HybPiper. Elliot, who was not at the meeting, provided the logo the very next day.
Development of HybPiper is ongoing, and I hope to maintain and extend the code to other applications in the future. For example, I’ve already received a few requests from users who would like to see an expansion of the handling of HybPiper for non-coding regions, such as plastid intergenic markers.